Pete Gordon asked a couple of questions regarding FDA regulations for Internet-based reporting software that interface with medical devices. The questions are essentially:
- How much documentation (SRS, SDS, Test Plan) is required and at what stage can you provide the documentation?
- How does the FDA view SaaS architectures?
The type of software you're talking about has no real FDA regulatory over site. The FDA has recently proposed new rules for connectivity software. I've commented on the MDDS rules, but Tim has a complete overview here: FDA Issues New MDDS Rule. As Tim notes, if the FDA puts the MDDS rules into place and becomes more aggressive about regulation, many software vendors that provide medical devices interfaces will be required to submit 510(k) premarket approvals.
Dealing with the safety and effectiveness of medical devices in complex networked environments is on the horizon. IEC 80001 (and here) is a proposed process for applying risk management to enterprise networks incorporating medical devices. My mantra: High quality software and well tested systems will always be the best way to mitigate risk.
Until something changes, the answer to question #1 is that if your software is not a medical device, you don't need to even deal with the FDA. The answer to question #2 is the same. The FDA doesn't know anything about SaaS architectures unless it's submitted as part of a medical device 510(k).
I thought I'd take a more detailed look at the architecture we're talking about so we can explore some of the issues that need to be addressed when implementing this type of functionality.
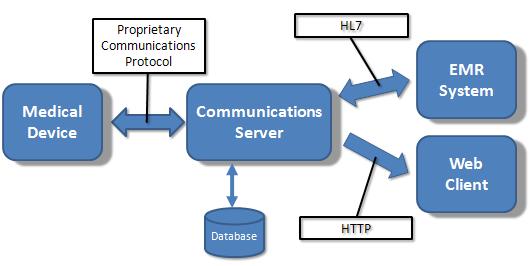
This is a simplified view of the way medical devices typically interface to the outside world. The Communications Server transmits and receives data from one or more medical devices via a proprietary protocol over whatever media the device supports (e.g. TCP/IP, USB, RS-232, etc.).
In addition to having local storage for test data, the server could pass data directly to an EMR system via HL7 or provide reporting services via HTTP to a Web client.
There are many other useful functions that external software systems can provide. By definition though, a MDDS does not do any real-time patient monitoring or alarm generation.
Now let's look at what needs to be controlled and verified under these circumstances.
- Communications interaction with proper medical device operation.
- Device communications protocol and security.
- Server database storage and retrieval.
- Server security and user authentication.
- Client/server protocol and security.
- Client data transformation and presentation to the user (including printed reports).
- Data export to others formats (XML, CSV, etc.).
- Client HIPAA requirements.
Not only is the list long, but these systems involve the combination of custom written software (in multiple languages), multiple operating systems, configurable off-the-shelf software applications, and integrated commercial and open source libraries and frameworks. Also, all testing tools (hardware and software) must be fully validated.
One of the more daunting verification tasks is identifying all of the possible paths that data can take as it flows from one system to the next. Once identified, each path must be tested for data accuracy and integrity as it's reformatted for different purposes, communications reliability, and security. Even a modest one-way store-and-forward system can end up with a hundred or more unique data paths.
A full set of requirements, specifications, and verification and validation test plans and procedures would need to be in place and fully executed for all of this functionality in order to satisfy the FDA Class II GMP requirements. This means that all of the software and systems must be complete and under revision control. There is no "implementation independent" scenario that will meet the GMP requirements.
It's no wonder that most MDDS vendors (like EMR companies) don't want to have to deal with this. Even for companies that already have good software quality practices in place, raising the bar up to meet FDA quality compliance standards would still be a significant organizational commitment and investment.
[…] Only medical device manufacturers have to be concerned with the FDA regulatory aspects of placing computing and networking components into a medical environment. I’ve previously discussed some of the regulatory and verification/validation issues with Connecting Computers to FDA Regulated Medical Devices. […]
[…] Its not the end of the world to be classified as a medical device, but verification and validation of these functions are not a trivial endeavor (see here). […]